
Theory and Simulation Facility
at Cornell University
Theory and Simulation are a critical part of the Materials by Design process employed by PARADIM. PARADIM’s user resources for theory and simulation are under the direction of Prof. Tomas Arias and located Cornell University. They are accessible remotely. All successful PARADIM projects should employ Theory and Simulation as an integral part of the research plan. Additional information on the use of these codes is available in the Materials by Design Toolbox
Computer cycles are available via clusters at Cornell University. In addition, PARADIM has received an allocation from XSEDE (an NSF supported supercomputer facility) and MARCC (through Johns Hopkins).
Capabilities and Highlights
Theory and Simulation New Capabilities
Materials-by-Design
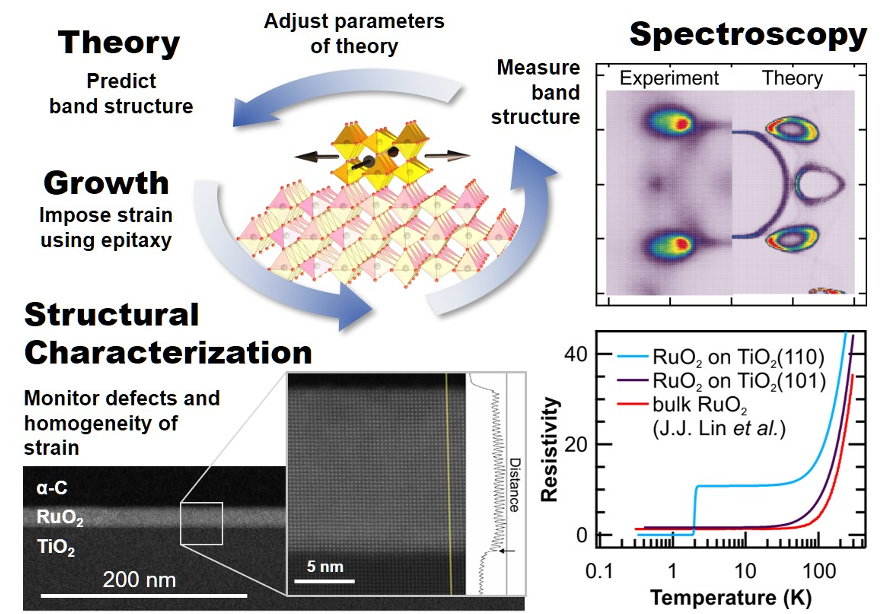
PARADIM Theory and Simulation Facility provides theoretical prediction of a material with desired properties.
Interface Quantum Materials
PARADIM developed Mismatched Interface Theory (MINT) allows the study of interface quantum materials theoretically.
Solid State and Quantum Chemistry Codes
JDFTx is a plane-wave density-functional theory (DFT) code designed to be as easy to develop with as it is easy to use. It is distributed under the GPL license (version 3 or higher) and publications resulting from its use must cite originating publication.
VASP is a computer program for atomic scale materials modeling, e.g. electronic structure calculations and quantum-mechanical molecular dynamics, from first principles. (Available through PARADIM Collaboration only)

Quantum Espresso is an Integrated suite of Open-Source computer codes for electronic-structure calculations and materials modeling at the nanoscale.

ABINIT is a program allows one to find the total energy, charge density and electronic structure of systems made of electrons and nuclei (molecules and periodic solids) within Density Functional Theory (DFT), using pseudopotentials and a plane wave or wavelet basis.

YAMBO is a FORTRAN/C code for Many-Body calculations in solid state and molecular physics. Yambo relies on the Kohn-Sham wave functions generated by two DFT public codes ABINIT and PWscf.

NAMD is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems.

DFTB+ is a fast and efficient versatile quantum mechanical simulation package. It is based on the Density Functional Tight Binding (DFTB) method, containing almost all of the useful extensions which had been developed for the DFTB framework so far.
CP2K is a quantum chemistry and solid state physics software package that can perform atomistic simulations of solid state, liquid, molecular, periodic, material, crystal, and biological systems.

SIESTA is both a method and its computer program implementation to perform efficient electronic structure calculations and ab initio molecular dynamics simulations of molecules and solids.
Classical molecular dynamics code, and an acronym for Large-scale Atomic/Molecular Massively Parallel Simulator.
Visualization Codes

VESTA is a 3D visualization program for structural models, volumetric data such as electron/nuclear densities, and crystal morphologies.
Molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting.
Software that can use to visualize Band structures obtain from VASP and plot graphs.
PARADIM Data Tools and Codes

Approved PARADIM Proposals gain access to the JHU-MARCC high-performance computing center

The SciServer PARADIM Data Collective (PDC) Cloud is a collaborative research platform for large-scale data-driven science. PARADIM users can run density functional theory (DFT) computational simulations and analyze microscopy data via inbuilt Jupyter notebook recipes.

PARADIM provides access to curated data sets, public data and private user data.
Materials Automated strives to automate repetitive data analysis workflows for anyone working with materials data in materials chemistry, physics, or science
e-Ptychography is a computational imaging technique developed at Cornell University by Zhen Chen and David Muller.
- e-Ptychography Repository
- Associated Publication - Cite if code is used

RigidRegistration is an image registration method optimized for low-SNR, cryogenic STEM data.
- Rigid Registration Repository
- Associated Publication - Must cite if code is used

vnc2flv is a remote desktop screen recorder.
Theory and Simulation Team

Dr. Tomás Arias
Leading the PARADIM Theory and Simulation Facility, Dr. Tomás Arias supports the Materials-by-Design process including developing tools JDFTx and MINT.
Sample Publications that Made Use of the Facility
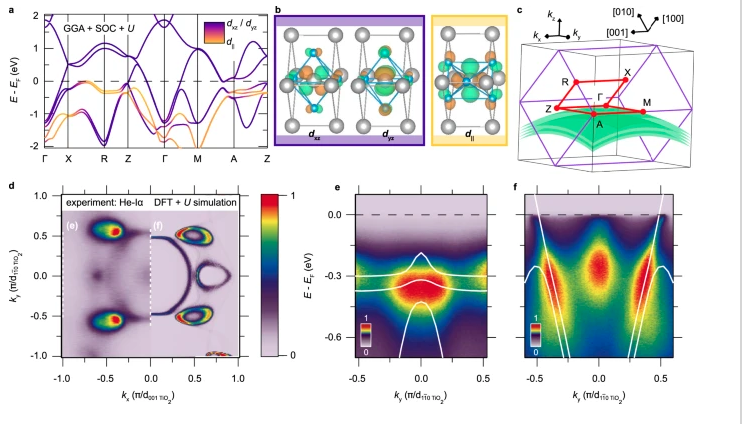
T. Yu, J. Wright, G. Khalsa, B. Pamuk, C.S. Chang, Y. Matveyev, X. Wang, T. Schmitt, D. Feng, D.A. Muller, G.H. Xing, D. Jena, and V. Strocov, "Momentum-Resolved Electronic Structure and Band Offsets in an Epitaxial NbN/GaN Superconductor/Semiconductor Heterojunction," Sci. Adv. 7, abi5833 (2021) and Highlight #53.
J.P. Ruf, H. Paik, N.J. Schreiber, H.P. Nair, L. Miao, J.K. Kawasaki, J.N. Nelson, B.D. Faeth, Y. Lee, B.H. Goodge, B. Pamuk, C.J. Fennie, L.F. Kourkoutis, D.G. Schlom, and K.M. Shen, “Strain-Stabilized Superconductivity,” Nat. Commun. 12, 59 (2021) and Highlight #36.
X. Zheng, E. Gerber, J. Park, D. Werder, O. Kigner, E.-A. Kim, S. Xie, and D.G. Schlom, "Utilizing Complex Oxide Substrates to Control Carrier Concentration in Large-Area Monolayer MoS2 Films," Appl. Phys. Lett. 118, 093103 (2021) and Highlight #37.
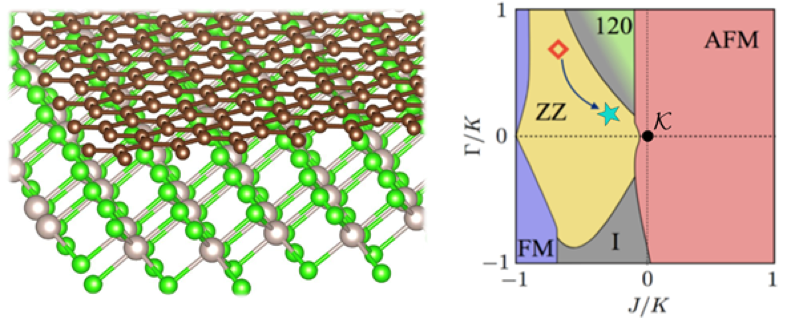
E. Gerber, Y. Yao, T.A. Arias, and E.-A. Kim, “Ab Initio Mismatched Interface Theory of Graphene on α−RuCl3: Doping and Magnetism,” Phys. Rev. Lett. 124, 106804 (2020) and Highlight #22.
Y. Ma, A. Edgeton, H. Paik, B. Faeth, C. Parzyck, B. Pamuk, S.-L. Shang, Z.-K. Liu, K.M. Shen, D.G. Schlom, and C.-B. Eom, “Realization of Epitaxial Thin Films of the Topological Crystalline Insulator Sr3SnO,” Adv. Mater. 32, 2000809 (2020) and Highlight #28.



